





Вход в систему
Опрос

Синдром Арнольда Киари
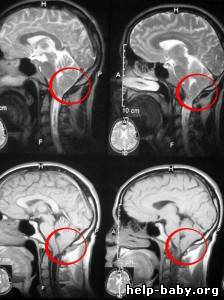
http://china-tcm.ru/content/sindrom-arnolda-kiari-simptomy-lechenie-diag...
Аномалия Арнольда-Киари – это врожденная патология развития ромбовидного мозга, проявляющаяся несоответствием размеров задней черепной ямки и мозговых структур находящихся в этой области, что приводит к опущению ствола головного мозга и миндалин мозжечка в большое затылочное отверстие и ущемлению их на этом уровне.
В большинстве случаев дефект сочетается с гидроцефалией и аномалиями развития спинного мозга. Причинами могут быть врождённая дисплазия (нарушение) широкого затылочного отверстия, размеры которого становятся значительно больше нормы.
Впервые он был описан Н. Chiari в 1896 г. Это состояние характеризуется каудальным смещением продолговатого мозга, моста и червя мозжечка, когда все эти структуры оказываются в шейной части позвоночника.
Частота этого заболевания составляет от 3.3 до 8.2 наблюдений на 100000 населения.
Истинная частота различных типов синдрома Арнольда - Киари, да и частота этого порока в целом, не установлены. Одной из причин отсутствия таких данных являются разные подходы к классификации этого порока. Согласно Международной классификации болезней, синдром Арнольда - Киари имеет отдельный шифр (Q07.0), однако определяется в ней как «... патологическое состояние, при котором происходит повышение внутричерепного давления в результате интракраниальной опухоли, окклюзионных форм гидроцефалии, воспалительного процесса, что в некоторых случаях приводит к вклинению мозжечка и продолговатого мозга в большое затылочное отверстие». В ультразвуковой пренатальной литературе до сих пор не удалось найти описаний случаев дородовой диагностики синдрома Арнольда- Киари, полностью соответствующих этим характеристикам.
Морфологические особенности различных типов порока Арольда - Киари определяют возможности пренатального выявления и прогноз для жизни.
Причины возникновения синдрома Арнольда- Киари до конца не установлены. Хромосомные аномалии при этой патологии, как правило, выявить не удается.
Патогенез (что происходит?) во время Аномалии Арнольда-Киари :
До настоящего времени патогенез патологии окончательно не установлен. По всей вероятности, этих патогенетических факторов три:
первый - наследственно обусловленные врожденные остеоневропатии,
второй - травматические повреждения клиновидно-решетчатой и клиновидно-затылочной части ската вследствие родовой травмы,
третий - гидродинамический удар ликвора в стенки центрального канала спинного мозга.
Анатомические особенности аномалии Киари
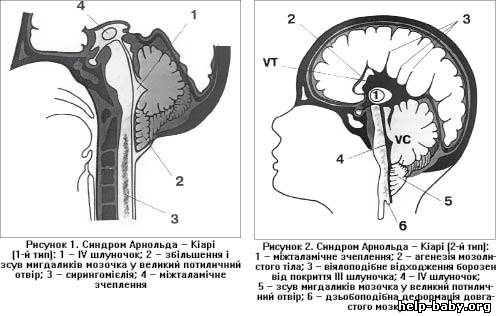
Мозжечок расположен в задней черепной ямке. (ЗЧЯ)
Миндалины - это нижняя часть мозжечка. В норме они расположены выше большого затылочного отверстия. При аномалии Киари миндалины мозжечка находятся ниже большого затылочного отверстия, в позвоночном канале.
Большое затылочное отверстие - это своеобразная граница между черепом и позвоночником, между головным и спинным мозгом. Выше большого затылочного отверстия находится задняя черепная ямка, ниже позвоночный канал.
На уровне большое затылочного отверстия нижний отдел ствола мозга (продолговатый мозг) переходит в спинной мозг. В норме спино-мозговая жидкость (ликвор) свободно циркулирует в субарахноидальных пространствах головного и спинного мозга. На уровне большого затылочного отверстия субарахноидальные пространства головного и спинного мозга соединяются, что обеспечивает свободный отток ликвора от головного мозга.
При аномалии Киари низко расположенные миндалины мозжечка затрудняют свободную циркуляцию спиномозговой жидкости между головным и спинным мозгом. Миндалины блокируют большое затылочное отверстие, как пробка затыкает бутылочное горлышко. В результате нарушается отток ликвора и развивается гидроцефалия.
Симптомы Аномалии Арнольда-Киари :
Киари (Chiari) выделил четыре типа аномалии с подробным их представлением. Данной классификацией врачи пользуются по настоящее время.
1. Аномалия Арнольда-Киари I типа представляет собой опущение структур ЗЧЯ в позвоночный канал ниже плоскости большого затылочного отверстия.
2. При аномалии Арнольда-Киари II типа - происходит каудальная дислокация нижних отделов червя, продолговатого мозга и IV желудочка, нередко развивается гидроцефалия.
3. Аномалия Арнольда-Киари III типа встречается редко, характеризуется грубым каудальным смещением всех структур задней черепной ямки.
4. Аномалия Арнольда-Киари IV типа - гипоплазия мозжечка без смещения его вниз.
Аномалии III и IV типов обычно несовместимы с жизнью.
Примерно у 80% пациентов аномалия Арнольда-Киари сочетается с патологией спинного мозга – сирингомиелией, которая характеризуется образованием в спинном мозге кист, вызывающих прогрессирующую миелопатию. Эти кисты образуются при опущении структур задней черепной ямки и сдавление шейного отдела спинного мозга.
Типичная клиническая картина аномалии Арнольда-Киари характеризуется следующими симптомами:
- боль в шейно-затылочной области усиливающаяся при кашле, чихании,
- снижение болевой и температурной чувствительности в верхних конечностях,
- снижение мышечной силы в верхних конечностях,
- спастичность верхних и нижних конечностей,
- обмороки, головокружения,
- снижение остроты зрения,
- в более запущенных случаях присоединяются: эпизоды апноэ (короткая остановка дыхания), ослабление глоточного рефлекса, непроизвольные быстрые движения глаз.
Возможные последствия, осложнения:
1. На фоне нарастающих признаков внутричерепной гипертензии (иногда без нее) отмечаются прогрессирующие нарушения функции мозжечка и сдавление шейного отдела спинного мозга, параличи черепных нервов.
2. Иногда аномалия Арнольда-Киари сочетается и с костными дефектами - окципитализацией атланта и базилярной импрессией (воронкообразное вдавление ската и краниоспинального сочленения).
3. Аномалии позвоночника, деформации стоп.
Диагностика Аномалии Арнольда-Киари :
Иногда аномалия Киари никак не проявляет себя и выявляется случайно при диагностических процедурах.
В настоящий момент методом выбора при диагностике данной патологии является МРТ головного мозга шейного и грудного отделов спинного мозга (для исключения сирингомиелии).
Лечение Аномалии Арнольда-Киари :
Если единственным симптомом заболевания является незначительной интенсивности болевой синдром, для лечения применяется консервативная терапия, которая включает в себя различные схемы с примененим нестероидных противовоспалительных препаратов и миорелаксантов.
При отсутствии эффекта от консервативной терапии в течение 2-3 месяцев или наличии у пациента неврологического дефицита (онемение, слабость в конечностях и т.д.) показано проведение операции.
Целью операции является– ламинэктомия, декомпрессивная краниоэктомия задней черепной ямки и пластика твёрдой мозговой оболочки. При подобной операции увеличивается объём задней черепной ямки и расширение затылочного отверстия, в результате чего прекращается сдавление нервных структур и нормализация тока цереброспинальной жидкости. В случаях сопуствующей гидроцефалии выполняется шунтирующая операция.
В Израиле больным предлагается щадящее и качественное лечение, которое после лечения позволяет больным вести полноценную жизнь. Хирургическое лечение синдрома Арнольда-Киари выполняется при помощи эндоскопа, при этом травмирующий эффект хирургического лечения сводится к минимуму. Метод минимально инвазивного хирургического лечения проводимый в израильских клиниках дает возможность пациентам с аномалией Арнольда-Киари вести в последующем полноценный образ жизни даже без лекарственной поддержки.
Симптоматология синдрома Арнольда-Киари
 Костноcyставные врожденные аномалии клинически менее очевидны сами по себе и гораздо более вследствие их тяжелых осложнений на центральной и периферической нервной системе. Неврологические проявления переносятся больным тяжелее всего и обусловливают неблагоприятное течение этого синдрома. Вообще, начало болезни медленное и нехарактерное.
Костноcyставные врожденные аномалии клинически менее очевидны сами по себе и гораздо более вследствие их тяжелых осложнений на центральной и периферической нервной системе. Неврологические проявления переносятся больным тяжелее всего и обусловливают неблагоприятное течение этого синдрома. Вообще, начало болезни медленное и нехарактерное.
Симптоматология, вначале очень стертая и даже отсутствующая долгое время, часто выявляется в результате вмешательства ряда разрешающих факторов, как, например, черепно-мозговая травма или инфекции носоглотки.
Первым проявлением, привлекающим внимание на наличие врожденной аномалии непосредственно после родов, является наличие миеломенингоцеле (спинномозговая грыжа). Позже отмечаются и другие клинические явления, указывающие на наличие костносустваных и неврологических аномалий, а именно:
- расщепление позвоночника,
- латеральное наклонение головы,
- девиация глазных яблок,
- головная боль и прерывистые или проходящие боли в области шеи (в особенности у детей старшего возраста и у взрослых), появляющиеся при движениях головы;
- тошнота, рвота.
У многих больных из-за блокады циркуляции спинномозговой жидкости между 4-м желудочком и цистернами основания черепа, уже в течение первых месяцев жизни, развивается иволютивная внутренняя гидроцефалия, обусловливающая появление очень многих и различных неврологических явлений. Постепенно череп ребенка увеличивается в размере и появляется затруднение в связи с кормлением, а также и дыхательные расстройства, а менингоцеле (когда оно существует) может изъязвиться.
Проявления внутричерепной гипертонии:
- сильные головные боли,
- папиллярный застой или атрофия зрительного нерва (поздняя),
- сопровождаемая прогрессивными нарушениями зрительной функции, вплоть до полной слепоты.
Мозжечковые проявления:
- головокружение;
- атаксия при ходьбе и в ортостатическом положении;
- дизартрия;
- нарушение глотания,
- интенционное дрожание,
- нистагм.
Проявления в сфере периферической нервной системы:
- парестезии, анестезии, парезы или параличи спастического типа,
- усиленные костносухожильные рефлексы,
- наличие рефлекса Бабинского.
Проявления в сфере черепно-мозговых нервов:
- односторонний или, реже, двусторонний паралич лицевого нерва;
- паралич глазодвигательных нервов, выраженный чаще всего внутренним косоглазием или диплопией.
Диагностика синдрома Арнольда-Киари .
Люмбальная пункция и биохимический и бактериологический анализы спинномозговой жидкости в большинстве случаев не доставляют значительных данных. Кроме того, применение люмбальной пункции может ухудшить состояние больного и даже вызвать смертельный исход ввиду внезапного понижения давления и полного проникновения мозжечковых миндалин и продолговатого мозга в позвоночный канал.
Обыкновенное рентгенологическое обследование выявляет следующие аспекты: малая задняя черепная ямка; расширение затылочного отверстия и позвоночного канала; гидроцефалия (большой череп с расхождением швов); отпечатки пальцев на костной пластинке черепа; сплющенность турецкого седла; шейное, дорзальное и люмбальное расщепление позвоночника.
Проведение дополнительных рентгенологических обследований (газовая миелоэнцефалография) противопоказаны у детей моложе 2 лет. Однако у детей старшего возраста и у взрослых они определяют косвенные и прямые признаки гидроцефалии, смещение продолговатого мозга и миндалин мозжечка, а также и сдавливание спинного мозга в шейной области.
Патологоанатомическое исследование. С анатомической и топографической точек зрения синдром Арнольда – Киари существует в виде четырех хорошо индивидуализированных типов, а именно:
- Первый тип, при котором существует растягивание и опущение миндалин мозжечка без смещения продолговатого мозга. Этот тип встречается чаще всего у детей старшего возраста и у взрослых. С клинической точки зрения он может оставаться всю жизнь бессимптомным и его выявление может произойти совершенно случайно.
- Второй тип, при котором смещается нижняя часть мозжечка и продолговатого мозга через большое затылочное отверстие в позвоночный канал. Этот тип встречается чаще у грудных детей и клинически проявляется гидроцефалией и зачастую и наличием миеломенингоцеле.
- Третий тип, с полным проникновением мозжечка в миеломенингоцеле шейных позвонков.
- Четвертый тип, при котором отмечается гипоплазия мозжечка, вызванная его тотальной грыжей; червь мозжечка невозможно различить, а миндалины и клочок мозжечка — едва заметны. Подобная форма встречается исключительно редко.
Помимо аномалии затылочной кости и позвонков, затрагивающих мозжечок и продолговатый мозг, существуют и другие черепно – позвоночные аномалии, а именно спаивание первого позвонка с затылочной костью, продвижение вверх позвоночника в черепную область из-за гипоплазии затылочной кости; слияние двух-трех позвонков (чаще всего 2-го и 3-го шейных позвонков), как это происходит при синдроме Klippel-Feil; шейное, дорсальное или люмбальное расщепление позвоночника.
Течение и прогноз синдрома Арнольда-Киари . Течение заболевания медленное. Появление синдрома у новорожденного несовместимо с его выживаемостью и его течение ведет быстро к летальному исходу.
В случае более медленного течения, синдром Арнольда – Киари осложняется появлением хронического арахноидита и паренхиматозными поражениями аксона, особо значимым это становится, когда наступает расстройство кровообращения в нервной ткани. Обычно, при подобных случаях нейропсихические расстройства отмечаются поздно и проявляются в форме параплегии, тетраплегии и запозданием психического развития.
Лечение синдрома Арнольда-Киари . Единственным аффективным лечением является хирургическое вмешательство. Показания к операции не ставятся на основании рентгенологического определения костных аномалий, а только тогда, когда последние сопровождаются тяжелыми неврологическими проявлениями. Хирургическое вмешательство заключается в затылочной краниотомии в сочетании с высокой ламинэктомией; твердая оболочка рассекается и оставляется открытой. Фиброзные спайки, часто обширные, существующие вокруг затылочного отверстия, пересекаются и отслаиваются.
При наличии выраженной гидроцефалии, производится деривация желудочка при помощи клапана Holter или Pudenz (по классическому методу хирургического вмешательства по поводу гидроцефалии).
Хотя в некоторых случаях, немедленные результаты и являются благоприятными, все же, все хирургические вмешательства при синдроме Арнольда – Киари связаны с большим послеоперационным риском, вследствие нарушений в области продолговатого мозга, которые могут наступить в ближайший послеоперационный период и вызывать часто смертельный исход.
- Блог пользователя - NanmuNan
- Для комментирования войдите или зарегистрируйтесь
Сейчас на сайте


Последние комментарии
3 года 21 неделя назад
3 года 47 недель назад
3 года 47 недель назад
4 года 5 недель назад
4 года 24 недели назад
4 года 25 недель назад
4 года 25 недель назад
4 года 27 недель назад
4 года 28 недель назад
4 года 30 недель назад